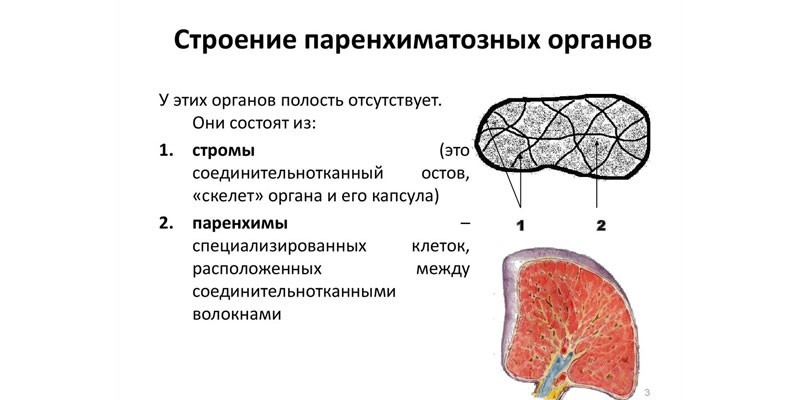
Синдром Фабрі є рідкісним генетичним захворюванням, при якому в організмі не вистачає ферменту альфа-G 4-галактозидази. Через це в клітинах різних органів та тканин накопичуються гліколіпіди. Частота народження варіюється в межах від 1:476000 до 1:117000.
Механізм розвитку
Інші назви синдрому Фабрі – дифузна універсальна ангіокератома, спадковий дистонічний ліпідоз, хвороба Андерсена. При такому захворюванні ліпідного обміну не відбувається повного розщеплення глікосфінголіпідів, через що вони накопичуються в наступних частинах організму:
- органи зору;
- ендотеліальні судини;
- гладком'язових клітинах;
- нирках;
- центральної нервової системи;
- серцевий м'яз.
Причини патології
Основна причина хвороби Фабрі – мутаційні зміни гена GLA. Він відповідає за кодування ферменту альфа-галактозидази А, який розташовується на довгому плечі статевої хромосоми Х. У його складі є 7 ділянок (екзонів) довжиною 92-291 пар основ.
З цього випливає, що синдром Фабрі – це Х-зчеплена хвороба. Вона відноситься до патологій з домінантним принципом успадкування з неповною проявністю у жінок. Механізм передачі хвороби:
- У жінки у хромосомному наборі є дві статеві хромосоми X, у чоловіка – X та Y.
- Під час зачаття дитина отримує від матері Х-хромосому, від батька – Х або Y. Від цього залежить статева приналежність малюка.
- Синдром Фабрі пов'язаний із Х-хромосомою. Її дитина отримує від матері чи батька. Жінка може передати мутацію як дочці, так і синові – в обох випадках ймовірність хвороби становить 50%. Від чоловіка дефектний ген може отримати лише дочку. Сини від батька із синдромом Фабрі народжуються здоровими.
- Хлопчику Х-хромосома з дефектом гена GLA може дістатися тільки від матері, оскільки від батька він отримує Y-хромосому. У такому разі дитина вважається гемізиготним носієм – у неї розвивається класичний тип синдрому Фабрі.
- Дівчатка можуть отримати дефектну Х-хромосому від матері чи батька. У такому разі дитина вважається гетерозиготним носієм, оскільки нормальний ген на другій хромосомі Х почасти компенсує мутацію. Через це хвороба протікає атипово: проявляється слабо, відрізняється пізнім початком.

Симптоми захворювання
Перша поява ознак цієї генетичної хвороби посідає раннє дитинство. Оскільки симптоми неспецифічні, діагноз найчастіше встановлюється після 10 років. З віком вираженість симптомів стає все яскравішою. У кожного пацієнта спостерігається суто індивідуальна клінічна картина.
Зовнішній вигляд хворих
До основних ознак синдрому Фабрі відносяться зміни зі боку шкірного покриву. Зовні у людини спостерігаються такі симптоми:
- Ангіокератоми. Є невеликими плямами від червоного до синюватого кольору. Новоутворення або плоскі, або трохи піднесені над шкірою.
- Висип. Локалізується на пальцях рук, сідницях, навколо пупка, в області паху, на колінах та ліктях. Обличчя уражається рідко, але висип може поширюватися на кон'юнктиву та ротову порожнину.
- Поступове збільшення плям. Це відбувається в міру дорослішання пацієнта.
- Незначне свербіння. Супроводжує плями, що з'явилися.
- Характерний фенотип. Зовні пацієнт нагадує хворого з акромегалією. У нього виступають лобові бугри і супраорбітальні дуги, збільшені губи, нижня щелепа, що виступає, запала перенісся. Згодом можливий розвиток деформацій міжфалангових суглобів стоп та кистей.

Особливості больового синдрому
Головна ознака хвороби – біль. Найчастіше вона виникає у віці 2 років, після чого поступово прогресує. Її викликають стреси, перегрів тіла, зміни погоди, втому та фізичні вправи. Синдром Фабрі супроводжують два види болю:
- Нейропатичний біль, або акропарестезія. Супроводжуються печінням і поколюванням у долонях та ступнях.
- Криз Фабрі. Це пекучий, сильний біль у долонях чи ступнях. Вона може поширюватися і інші частини тіла.
Неврологічні прояви
Синдром Фабрі супроводжується численними неврологічними порушеннями. Через накопичення сфінголіпідів порушується мозковий кровообіг. Наслідком цього стають такі симптоми:
- уповільнення розумового процесу;
- геморагічний чи ішемічний інсульт;
- судоми;
- порушення мови;
- посилене потовиділення;
- проблеми із пам'яттю;
- зміни поведінки;
- порушення координації;
- паралічі, парези.
Тактика лікування та прогноз
Оскільки хвороба є генетичною, її повністю вилікувати не можна. Прогноз синдрому Фабрі умовно сприятливий. При постійній замісній терапії вдається запобігти розвитку небезпечних наслідків та покращити якість життя хворого. Якщо у пацієнта розвиваються ускладнення, більшість випадків закінчуються летальним результатом.
Основний метод лікування хвороби – ферментозаміщення за допомогою довічного прийому препаратів, що містять альфа-галактозидазу А. Найчастіше використовувані ліки:
- Фабразіма. Це агалсідаза-бета, яка синтезується із яєчників китайських хом'яків.
- Реплагала. Це агалсідаза-альфа, що виробляється на основі фібробластів людської шкіри.

Медикаменти вводяться внутрішньовенно у дозуванні, суворо визначеному лікарем залежно від індивідуальних особливостей пацієнта. Стандартна схема – внутрішньовенне введення 2 рази на місяць. Додатково пацієнту призначають ліки, що зменшують симптоми захворювання:
- антиконвульсанти;
- кортикостероїди;
- нестероїдні протизапальні препарати;
- знеболювальні;
- антигіпертензивні засоби;
- інгібітори АПФ;
- імуномодулятори;
- блокатори рецепторів ангіотензину